T=1.0D NOINTER GNORM=0.1 PM3 GEO-OK : 1行目 ← キーワード
CH4 : 2行目 ← コメント行
comments : 3行目 ← コメント行
C 0.000 0 0.000 0 0.000 0 0 0 0 : 4行目 ← 内部座標
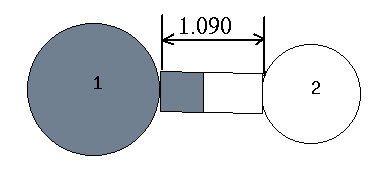
H 1.090 1 0.000 0 0.000 0 1 0 0 : 4行目 ← 内部座標
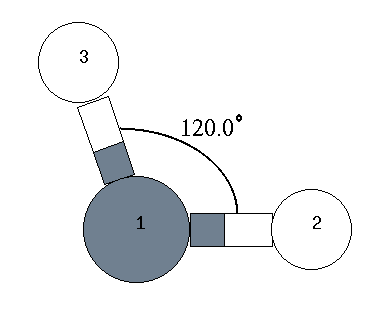
H 1.090 1 109.000 1 0.000 0 1 2 0 : 4行目 ← 内部座標
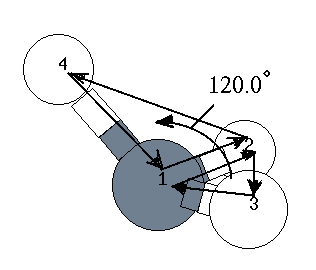
H 1.090 1 109.470 1 120.000 1 1 2 3 : 4行目 ← 内部座標
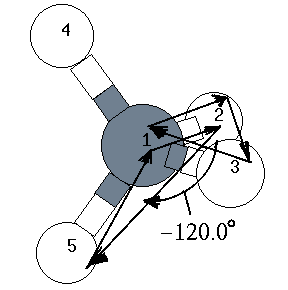
H 1.090 1 109.470 1 -120.000 1 1 2 3 : 4行目 ← 内部座標
: 5行目 ← 空行
1 最適化する。 0 初期値のまま最適化しない。 -1 反応座標とする。(難しいので、ここでは説明しない。)である。 最初は何も考えずに例の様に参照する原子の無い所は 0 にし、 それ以外は 1 にすれば良い。
そして、5行目の何もない空行を入れて、定義終了とする。
| 番号 | 元素記号 | 結合している元素との距離 | 結合角の大きさ | 2面角の大きさ | 結合している元素の番号 | 結合角を作る元素の番号 | 2面角を作る元素の番号 |
|---|---|---|---|---|---|---|---|
| 1 | C | 0.00 | 0.00 | 0.00 | 0 | 0 | 0 |
| 2 | H | 1.090 | 0.00 | 0.00 | 1 | 0 | 0 |
| 3 | H | 1.090 | 109.00 | 0.00 | 1 | 2 | 0 |
| 4 | H | 1.090 | 109.00 | 120.00 | 1 | 2 | 3 |
| 5 | H | 1.090 | 109.00 | -120.00 | 1 | 2 | 3 |
| 6 |
また、入力データの例を載せるので、これを真似ながら作成するのもいいでしょう。 データの作成は、慣れと真似が大切です。
このページのトップへ戻る
ハミルトニアンの指定 ハミルトニアンの指定何も入れないとMNDO 法を使用する。 MINDO/3 : MINDO/3 法を使用。 AM1 : AM1 法を使用。 PM3 : MNDO-PM3 法を使用。おすすめ。 計算を早く収束させる為のキーワード SHIFT = n : 計算開始に減衰ファクターn を定義する。 PULAY : Pulay の強制収束法を使用する。 CAMP : CAMP-KING を用いた収束法。 その他、よく使っているキーワード T=n : 最大CPU 時間 n 秒。入れなくても約 24時間で終了。 NOINTER : 原子間距離を印字しない。 GNORM=n.n : エネルギーの勾配ノルムがn.n以下になったら、計算終了。1.0〜0.1を推奨。 GEO-OK : 原子が異常に接近した場合のチェックを無視。 UHF : 非制限Hartree-Fock計算。入れないと、Hartree-Fock(RHF)計算を行う。その他の使用したことのあるキーワードを、 「研究室用ページ」 に載せてあります。 多くのキーワードがありますが、難しいので勉強してから使いましょう。
このページのトップへ戻る
% which mp.exe (名称は、これ以外かも知れません。) % which xmol上のように出ればOKです。
そして、入力ファイルのあるディレクトリに移動し、計算を実行する。 この時、入力ファイルの ".dat" は入れないこと。入れると計算できません。
% ls filename.dat % mp.exe filename負荷をかけすぎないため、同時に計算をするのは止めましょう。
計算中及び、終了後に次のような拡張子を持ったファイルが出力されます。 それぞれの意味は次のようになります。
計算中、終了後にあるファイル
filename.out : 結果出力ファイル
filename.res : 再起動用ファイル
filename.den : 密度行列ファイル
filename.log : 計算経過モニターファイル
filename.end : 停止命令用ファイル
"filename.res"と"filename.den"は、計算中約1時間事に作成される。
そして、計算をやり直す時に両方とも必要で、キーワード"RESTART"
を入れ計算を行うと、途中から計算を始めることが出来る。
計算中のみあるファイル
filename.temp : 入力ファイルのコピー ( 計算中に"filename.dat"を書き換えても良い)
tmp.FAAa002.d : 不明。
これらのファイルは強制的に計算を終了させた場合消えずに残っているため、
ファイル容量の無駄な為、消すこと。
計算後に出力されるファイル
filename.arc : 結果の要約ファイル
同じ"filename"で計算を行うと、このファイルのみ上書きされずに
"filename.arcaa" → "filename.arcab" → …
となる。そして、"xmol" で結果の構造を確認する時は、このファイルを読み込む。
このページのトップへ戻る
SUMMARY OF PM3 CALCULATION
MOPAC 93.00
C H4
T=1.0D NOINTER GNORM=0.1 PM3 GEO-OK ←(入力したキーワード)
CH4 ←(入力したコメント)
comments ←(入力したコメント)
PETERS TEST WAS SATISFIED IN BFGS OPTIMIZATION
SCF FIELD WAS ACHIEVED
↑
(計算結果の説明。"TKAE CARE" などが出ている時は、"filename.out"のファイルを
良く見る必要がある。計算が正しく終了していない場合がある。)
HEAT OF FORMATION = -13.025645 KCAL = -54.49930 KJ
↑(生成エネルギー。低い方安定な構造である。)
ELECTRONIC ENERGY = -384.912477 EV
CORE-CORE REPULSION = 204.376508 EV
DIPOLE = 0.00001 DEBYE SYMMETRY: Td
NO. OF FILLED LEVELS = 4
IONIZATION POTENTIAL = 13.642261 EV
HOMO LUMO ENERGIES (EV) = -13.642 4.245
MOLECULAR WEIGHT = 16.043
SCF CALCULATIONS = 5
COMPUTATION TIME = 0.060 SECONDS
FINAL GEOMETRY OBTAINED CHARGE
↓(最適化された構造と、それぞれの原子の電荷。ここから下の部分を残してファイル名を
"filename.dat"にすればこの構造から計算を始めることが出来る。この時、"CHARGE"の
数値を消す必要はない。)↓
T=1.0D NOINTER GNORM=0.1 PM3 GEO-OK
CH4
comments
C 0.00000000 0 0.0000000 0 0.0000000 0 0 0 0 -0.1104
H 1.08702831 1 0.0000000 0 0.0000000 0 1 0 0 0.0276
H 1.08695850 1 109.4715814 1 0.0000000 0 1 2 0 0.0276
H 1.08692930 1 109.4620522 1 120.0000000 0 1 2 3 0.0276
H 1.08695094 1 109.4687338 1 -120.0000000 0 1 2 3 0.0276
このページのトップへ戻る
*******************************************************************************
** MOPAC 93 (c) Fujitsu **
*******************************************************************************
PM3 CALCULATION RESULTS
*******************************************************************************
* MOPAC 93.00 CALC'D. Thu Jun 11 19:45:36 1998
* GEO-OK - OVERRIDE INTERATOMIC DISTANCE CHECK
* T= - A TIME OF 1.000 DAYS REQUESTED
* DUMP=N - RESTART FILE WRITTEN EVERY 3600.000 SECONDS
* PM3 - THE PM3 HAMILTONIAN TO BE USED
* NOINTER - INTERATOMIC DISTANCES NOT TO BE PRINTED
* GNORM= - EXIT WHEN GRADIENT NORM DROPS BELOW .100
***********************************************************************080BY080
↑(キーワードの説明)↑
T=1.0D NOINTER GNORM=0.1 PM3 GEO-OK
CH4
comments
ATOM CHEMICAL BOND LENGTH BOND ANGLE TWIST ANGLE
NUMBER SYMBOL (ANGSTROMS) (DEGREES) (DEGREES)
(I) NA:I NB:NA:I NC:NB:NA:I NA NB NC
1 C
2 H 1.09000 * 1
3 H 1.09000 * 109.00000 * 1 2
4 H 1.09000 * 109.00000 * 120.00000 1 2 3
5 H 1.09000 * 109.00000 * -120.00000 1 2 3
↑(入力データ)↑
↓(入力データのxyz座標)↓
CARTESIAN COORDINATES
NO. ATOM X Y Z
1 C 0.0000 0.0000 0.0000
2 H 1.0900 0.0000 0.0000
3 H -0.3549 1.0306 0.0000
4 H -0.3549 -0.5153 -0.8925
5 H -0.3549 -0.5153 0.8925
H: (PM3): J. J. P. STEWART, J. COMP. CHEM. 10, 209 (1989).
C: (PM3): J. J. P. STEWART, J. COMP. CHEM. 10, 209 (1989).
MOLECULAR POINT GROUP : C1
RHF CALCULATION, NO. OF DOUBLY OCCUPIED LEVELS = 4
CYCLE: 1 TIME: 0.012 TIME LEFT: 24.00H GRAD.: 0.573 HEAT:-13.02545
TEST ON GRADIENT SATISFIED
PETERS TEST SATISFIED
↓(ここから計算結果の内容)↓
-------------------------------------------------------------------------------
T=1.0D NOINTER GNORM=0.1 PM3 GEO-OK
CH4
comments
PETERS TEST WAS SATISFIED IN BFGS OPTIMIZATION
SCF FIELD WAS ACHIEVED
PM3 CALCULATION
MOPAC 93.00
FINAL HEAT OF FORMATION = -13.02565 KCAL = -54.49930 KJ
TOTAL ENERGY = -180.53597 EV ←(最適化された分子の全エネルギー)
ELECTRONIC ENERGY = -384.91248 EV POINT GROUP: Td
CORE-CORE REPULSION = 204.37651 EV
↑(電子エネルギーと、核間エネルギー)↑
IONIZATION POTENTIAL = 13.64226
NO. OF FILLED LEVELS = 4
MOLECULAR WEIGHT = 16.043
SCF CALCULATIONS = 5
COMPUTATION TIME = 0.054 SECONDS
↓(最適化構造)↓
ATOM CHEMICAL BOND LENGTH BOND ANGLE TWIST ANGLE
NUMBER SYMBOL (ANGSTROMS) (DEGREES) (DEGREES)
(I) NA:I NB:NA:I NC:NB:NA:I NA NB NC
1 C
2 H 1.08703 * 1
3 H 1.08696 * 109.47158 * 1 2
4 H 1.08693 * 109.46205 * 120.00000 1 2 3
5 H 1.08695 * 109.46873 * -120.00000 1 2 3
MOLECULAR POINT GROUP : Td ←(構造の対象性の属する群。群論を学んでから)
EIGENVALUES ←(分子軌道エネルギー)↓
-29.88062 -13.64326 -13.64292 -13.64226 4.24497 4.59401 4.59449 4.59468
NET ATOMIC CHARGES AND DIPOLE CONTRIBUTIONS ←(原子上の電荷)
↓
ATOM NO. TYPE CHARGE ATOM ELECTRON DENSITY
1 C -0.110420 4.1104
2 H 0.027593 0.9724
3 H 0.027612 0.9724
4 H 0.027605 0.9724
5 H 0.027611 0.9724
DIPOLE X Y Z TOTAL
POINT-CHG. 0.000 0.000 0.000 0.000
HYBRID 0.000 0.000 0.000 0.000
SUM 0.000 0.000 0.000 0.000
CARTESIAN COORDINATES ←(xyz座標)↓
NO. ATOM X Y Z
1 C 0.0000 0.0000 0.0000
2 H 1.0870 0.0000 0.0000
3 H -0.3623 1.0248 0.0000
4 H -0.3621 -0.5124 -0.8875
5 H -0.3623 -0.5124 0.8875
ATOMIC ORBITAL ELECTRON POPULATIONS ←(軌道の電子密度)
↓
1.11082 0.99987 0.99987 0.99986 0.97241 0.97239 0.97240 0.97239
TOTAL CPU TIME: 0.06 SECONDS
== MOPAC DONE ==
NO ATOMS IN SYSTEM
このページのトップへ戻る
% xmol &
このページのトップへ戻る
コメントまたはアドバイスなどがあれば以下のアドレスへどうぞ。
 yagi@tube.ee.uec.ac.jp
yagi@tube.ee.uec.ac.jp