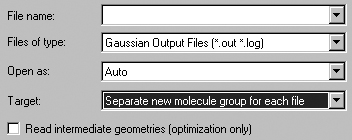
Figure 22. GaussView Fields in the Open File Dialog
GaussView adds three fields and a checkbos to the standard Open File dialog provided by the operating system.
In this section, we consider the GaussView features for opening model files and other types of molecule specification input (e.g., PDB files and Gaussian input files).
The File=>Open menu item and the Open button both bring up the Open File dialog. The format of this dialog follows the native form for the local computer, with the addition of some GaussView-specific fields which are illustrated in Figure 22.
Figure 22. GaussView Fields in the Open File Dialog
GaussView adds three fields and a checkbos to the standard Open File dialog
provided by the operating system.
These additional items have the following meanings. The Files of Type popup is used to select the file type to open. It also acts as a filter selecting which files are displayed in the dialog's file list. The file list will be limited to the files with the selected extension(s). Supported file types include:
Gaussian Input Files (.com or .gjf)
Gaussian Output Files (.log or .out)
Cube Files (.cub)
Gaussian Checkpoint Files (.chk)
Gaussian Formatted Checkpoint Files (.fch*)
Brookhaven PDB Files (.pdb or .ent)
MDL Mol Files (.mol)
Sybyl Mol2 Files (.mol2 or .ml2)
CIF Files (.cif)
All files may also be displayed if desired by selecting the All Files item.
The Open as popup is used to force an input file to be interpreted as the specified file type, regardless of the actual file extension. All supported file types except Gaussian cube files are available as choices here. The default is Auto, which identifies the file type by its extension alone.
The Target popup specifies where the read-in structure should be placed:
Separate new molecule group for each file: Create a new, one member molecule group for each file that is read in. This is the default.
Single new molecule group for all files: Create one new molecule group, and make each file a model within it. When opening a single file, this is equivalent to the first option.
Append all files to active molecule: Place the structure(s) from all opened file into the current model. This will result in additional fragments being added to the model.
Add all files to active molecule group: Add each file as a separate new model within the current molecule group. The current model/molecule is left unchanged.
In general, only the first structure present in a file is input. Thus, only the structure in the first job step of Gaussian input files is retrieved. However, when selected, the Read intermediate geometries (optimization only) checkbox causes GaussView to retrieve all geometries that are present in some Gaussian results files as separate models within the designated target. This box applies to results files from geometry optimizations, IRC jobs, ADMP and BOMD trajectory calculations and potential energy surface scans: all jobs with an optimization component. It is not valid with the Append all files to active molecule choice.
Pressing the View File button or selecting the Results=>View File menu item opens the file which was opened for the current model, provided that the molecule has not been structurally modified in any way since it was opened. In the case of binary input files like Gaussian checkpoint files, the displayed file will be the one that was created automatically by GaussView with the formchk utility.
The File=>Recent Files menu path contains a list of files that have been opened most recently. You can specify the number of items in the list with the Maximum Recent Files file in the File/Directory preferences. However, using these shortcuts always reads in only a single geometry from the specified file into a new model group, regardless of the current setting of the current settings in the Open File dialog or the method that was used to open the file on the previous occasion. The file list is saved across GaussView sessions.
Previous Next