Special Considerations for Various Gaussian Job Types
This section summarizes information about setting up various Gaussian
job types for which some special steps are required All GaussView features
mentioned are discussed in detail earlier in this book.
Transition Structure Optimizations: Opt=QST2 and Opt=QST3
Gaussian STQN-based transition structure optimizations require two or
three structures as input. To set up these jobs, you must create a molecule
group containing the required number of structures. If you plan on running an
Opt=QST3 job, then the transition structure initial guess should be model
3.
Once you have done so, the TS (QST2) and TS (QST3) options will
be enabled in the Optimize to a field for the Optimization job
type in the Job Type panel of the Gaussian Calculation Setup dialog.
Verifying and Specifying Atom Equivalencies
In most cases, GaussView will automatically identify the corresponding
atoms in the multiple structures for these transition state optimizations. However,
you can verify this using the Connection Editor, accessed via the Connection
Editor button on the toolbar or the Edit=>Connection Editor menu item.
Calculations on Polymers, Surfaces and Crystals
You can set up jobs for Gaussian's Periodic Boundary Conditions facility
using the Crystal Editor (reached via the Crystal Editor button or the
Edit=>PBC menu path). Once you have defined a unit cell, GaussView
automatically sets up PBC jobs for this structure by including the translation
vectors within the molecule specification. This is indicated by the enabling
of the PBC panel in the Gaussian Calculation Setup dialog, and
the checked Use PBC item. Note that for normal cases, you do not need
to access this panel at all and can proceed directly to setting up Gaussian
input in the normal manner.
Multi-Layer ONIOM Calculations
GaussView contains several features for setting up ONIOM calculations.
Assigning Atoms to Layers
The Layer Editor allows you to graphically assign atoms to various ONIOM
layers. It is accessed via the toolbar's Select Layer button or via the Edit=>Select
Layer menu item.
Assigning Molecular Mechanics Atoms Types
GaussView will assign Molecular Mechanics atoms types for UFF, Dreiding
and Amber (including Amber charges) to all atoms in the molecule automatically.
You can view and modify these using the Atom List Editor (reached via
the Atom List Editor button on the toolbar or the Edit=>Atom List menu path).
Defining Link Atoms
GaussView will automatically assign minimal link atom information for
the appropriate atoms in an ONIOM calculation. However, these are always handled
in the same way and may require modification for your purposes. Link atoms generated
by GaussView are always hydrogens (using the H_ UFF and Dreiding atom
types and the HR Amber atom type, where R is the element of the
linked-to atom). The only other link atom parameters which is included is the
linked-to atom (the atom in the higher layer to which the current atom is bonded);
all other parameters are left blank.
Specifying the Model Chemistry for Each Layer
Once you have prepared the structure and specified all necessary parameters,
you can set up an ONIOM calculation via the Method panel of the Gaussian
Calculation Setup dialog. The Multilayer ONIOM Model checkbox indicates
that this will be an ONIOM calculation (see Figure 55).
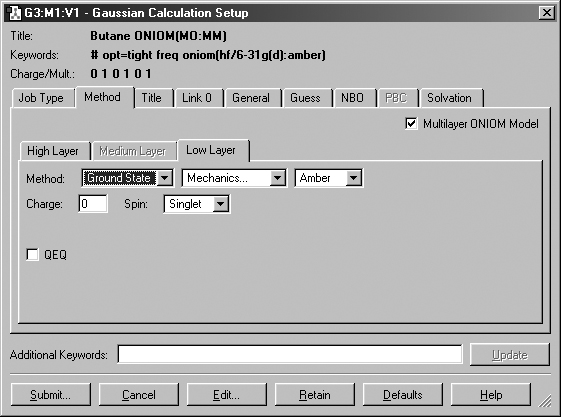
Figure 55. Setting Up a Gaussian Input File for an ONIOM Job
This example is preparing an input file for a two-layer ONIOM calculation.
When Multilayer ONIOM Model is checked, the additional tabs appear in the Method
panel. Each of them allows you to specify the theoretical method and basis set
for the corresponding layer. In this case, we are using the Amber Molecular
Mechanics method for the Low layer.
Specifying CASSCF Active Spaces Using Guess=Permute
GaussView can make it easy to specify CASSCF active space. The MOs
dialog allows you to generate, view, select and reorder the starting orbitals.
It is reached with the Edit=>MO Editor menu path and via the MO Editor button
on the toolbar.
Modifying Redundant Internal Coordinates (Geom=ModRedundant)
You can specify additions and other modifications to redundant internal coordinates
for geometry optimizations and other jobs by using the Redundant Coordinate
Editor, reached via the Redundant Coordinate Editor button on the toolbar
or the Edit=>Redundant Coord Editor menu path.
Previous Next